
There is a quote that states: ‘A couple of days in the laboratory can save a couple of hours in the library’. This sentiment once typified the attitude of a lot of scientists, but those days are over. It may not have reached the point where a couple of hours on the computer can save a couple of days in the laboratory, but it’s heading that way. The laboratory is part of a business process and, as such, it is subject to the same productivity and efficiency targets that apply to other parts of the business.
Most laboratory informatics projects focus on the return on investment, typically quantified by streamlining data input through the elimination of bottlenecks, by interfacing systems, and by removing manual processes involving paper. Most projects will also specify long-term gains through establishing a knowledge repository, but this is where quantitation becomes difficult. It’s not unusual in the early days of an informatics deployment, for example, for a simple search to uncover prior work that can save reinventing the wheel. But, as time goes on, it becomes the norm to check before starting an experiment. There is, however, another side to exploiting the knowledge base – which we’re only just starting to come to terms with. The need to delve deeper into the knowledge base to visualise and interpret relationships and correlations is growing. ‘Big data’ is the popular term being assigned to the data problem, as it applies to all walks of life. This puts the emphasis on ensuring that we have confidence in the integrity, authenticity, and reliability of the data going in, and that the appropriate tools are available to search, analyse, visualise, and interpret the information coming out.
These requirements are driven by the requirements of the laboratory’s customers for robust, reliable, and meaningful scientific information and data that is delivered in a timely and cost-efficient way. The time-honoured principles of the scientific method provide the basis for the integrity, authenticity, and reliability of scientific data, but those principles need to be reinforced in the context of regulatory compliance and patent evidence creation.
Productivity/business efficiency
The basic objective in deploying laboratory informatics systems is to improve laboratory productivity and business efficiency. To maximise the benefits, it is important to consider the wider laboratory and business processes that may be affected by the new system. It is easy to fall into the trap of just ‘computerising’ an existing laboratory function, rather than looking at the potential benefits of re-engineering a business process. The use of tools such as 6-Sigma or Lean can help considerably. Nevertheless, it is prudent to be careful with the use of these tools, depending on the nature of the lab. For example, high-throughput, routine-testing laboratories, which basically follow standard operating procedures, are more receptive to process improvement. Discovery/research laboratories however, which are less structured and are dependent on more diverse and uncontrolled processes, are less likely to benefit from formal process re-engineering.
Productivity and business efficiency are usually measured in financial terms, although this may be translated into time-savings or, in some cases, the numbers of tests, samples, experiments completed. It is necessary, therefore, to be able to quote ‘before and after’ figures for any deployment project. Establishing a baseline metric is an important early step in the project.
The tools can facilitate improvement through well thought-out deployment, but also offer the capability to monitor and improve processes.
Costs/return on investment
Any organisation considering the implementation of a new informatics or automation system will want to investigate the return on investment (ROI), or cost/benefit. This is usually extremely difficult, since many of the projected benefits will be based on a certain amount of speculation and faith. However, there are some important points to consider in building the cost/benefit case. The costs associated with managing paper-based processes (e.g. notebooks, worksheets, etc.) through their full lifecycle in the lab are not always fully visible or understood.
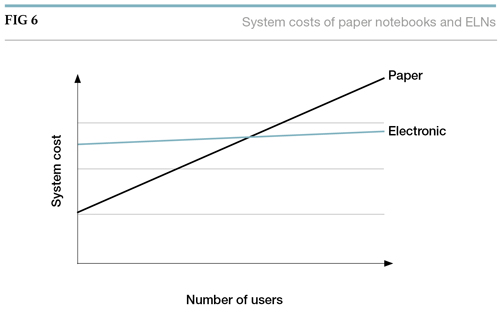
Apart from the material costs, and the costs of the archive process, there is a hidden cost – and the time taken in writing by hand, cutting, pasting, transcribing, and generally manipulating paper, as well as approval and witnessing processes, all contribute to this hidden cost. It is normal in building the cost/benefit equation to look at how much of a scientist’s time is spent managing the paper-based processes, and to use this as a basis for potential time-savings with an electronic solution (see Figure 6). Although the start-up costs are high for an electronic solution, the incremental cost of adding new users and increasing storage space is modest.
ROI tends to focus on the short term: how soon can one get a return on the money invested in deploying a new system? But the true value of the system may be in the long term and, therefore, far more difficult to measure as the value will be determined by behavioural changes. There is a growing body of evidence being presented at conferences on electronic laboratory notebooks (ELNs) by numerous companies that have implemented them, showing that the short-term time savings associated with the electronic solution are significant. These organisations also list a number of other non-quantifiable, long-term benefits such as:
- Scientists spending more time in the laboratory;
- It is easier to find information in a searchable archive;
- It is easier to share information;
- Increased efficiency through the elimination of paper;
- A reduced need to repeat experiments (knowingly or unknowingly);
- Improved data quality;
- A smooth transition when people leave the company; and
- Online use in meetings.
Regulatory compliance
The early research phases in the pharmaceutical industry comprises the testing of large numbers of chemicals to see if any of them have potential as a new drug. Only the best will go on to more extensive testing. There has been a ‘consensus’ that regulatory work does not start until the chemical has been chosen. Then adherence to GMP[3] (good manufacturing practice) and GLP[4] (good laboratory practice) starts, and the IT systems need to be in compliance with the local requirements for IT systems. In the US, this is 21 CFR Part 11[5] and in the EU it is GMP Annex 11.[6] While this may be at least partially correct, the fact is that the data, and of course the IT systems that hold the data, need to be under control for another business reason: patents.
The US patent system is based on ‘First to Invent’, and that means it must be possible to prove the date of the invention. Traditionally, this has been done using bound paper notebooks, where the entries have been dated and signed, and co-signed by a witness. Paper notebooks can be admitted as evidence if they can be demonstrated to be relevant. Electronic records are equally relevant, as the judgment is made on the evidence – not on the medium that holds the evidence. One important factor is the data integrity, which must be possible to prove in court if necessary. Bound paper logbooks are still being used to a large extent, as most legal advisors don’t feel comfortable with electronic data. It may be smart to talk to patent lawyers before starting to create electronic lab data.
How can we prove that the IT system is good enough?
The answer is, of course, validation. Actually, validation of processes is nothing new; that has been a part of the GMP and GLP regimes since they were introduced. An IT system is a part of the process and must therefore be validated as well.
The industry asked the US Food and Drug Administration (FDA) how it would handle electronic signatures, and accordingly 21 CFR Part 11 saw the light in 1997. The surprise was that most of the two-page document was about electronic data, and only a little about signatures.
This, however, does make sense. How can scientists use an E-signature if they are not sure that the data is (and will be) valid? They can’t; they need to have control of your data before they can sign it electronically. The EU also came up with an equivalent to 21 CFR Part 11, namely the EU GMP Annex 11. This was revised in 2011 but does not improve on the first version. But a really good document covering electronic data and signatures is yet another document numbered 11, the PIC/S PI 011.[7] This is a 50-page document with the same requirements as Part 11, but it includes also a lot of explanations. PIC/S is the organisation for European pharma inspectors. They do stress that this document is not a regulatory requirement, only an explanation to the inspectors on how to handle IT systems. How that cannot be a requirement document, is hard to understand, however. The main difference between Part 11 and Annex 11 is that the latter also includes risks. IT validation shall be based on risks; high-risk systems need more validation than low-risk systems.[8]
This follows the same line of thought that the FDA started in the early 2000s: know your processes, and base the work on the risks they encompass.

How do we validate our IT system?
The best answers are in a guidebook called GAMP5[9]. GAMP also has a few more detailed sub-books.
What is written in this guide to a smart laboratory is of course just an overview. Please see GAMP5 for more details.
The GAMP5 way of validating the IT systems is as follows:
- Risk management to decide how important the system is in the process;
- Categories of software to decide what needs to be done;
- Combination of risks and categories to decide what to do for this system; and
- Testing guide for how to test the system.
Risk management
Identify regulated E-records and E-signatures:
- Is the record required for regulatory purposes? Is it used electronically? Is a signature required by GMP/GLP/GCP?
Assess the impact of E-records:
- The classification of potential impact on patient safety and/or product quality: is it high/medium/low?
Assess the risks of E-records:
- The impact and likelihood/probability of problems being detected/happening: is it high/medium/low?
Implement controls to manage risks:
- Modify processes, modify the system design; apply technical or procedural controls.
Monitor effectiveness of controls:
- Verify effectiveness, consider if unrecognised hazards are present; assess whether the estimated risk is different and/or the original assessment is still valid.
GAMP5 software categories
Category 1: Infrastructure software
- Definition: layered software (i.e. upon which applications are built). Software used to manage the operating environment.
- Example: operating systems, database engines, statistical packages, programming languages.
- Validation: record version and service pack. Verify correct installation.
Category 2: This category is no longer in use
Category 3: Non-configured products
- Definition: off-the-shelf solutions that either cannot be configured or that use default configuration. Run-time parameters may be entered and stored, but the software cannot be configured to suit individual business processes.
- Example: firmware-based applications, COTS, instruments.
- Validation: the package itself. Record version and configurations, verify operations against user requirements. Consider auditing the vendor. Risk-based tests of application: test macros, parameters, and data integrity.
Category 4: Configured products
- Definition: software, often very complex, that can be configured by the user to meet the specific needs of the business process. Software code is not altered.
- Example: LIMS/SCADA/MES/MRP/EDMS/clinical trials, spreadsheets and many others (See GAMP5).
- Validation: life cycle approach. Risk-based approach to supplier assessment and other testing. Record version and configuration, verify operation against user requirements. Make sure SOPs are in place for maintaining compliance and fitness for\ intended use, as well as for managing data.
Category 5: Custom applications
- Definition: software, custom-designed and coded to suit the business processes.
- Example: it varies, but includes all internally/externally developed IT applications, custom firmware and spreadsheet macros. Parts of Category 4 systems may be in this category.
- Validation: the same as Category 4, plus more rigorous supplier assessment/audit, full life cycle documentation, design and source code review.
System validation
The validation itself usually needs to be divided into more manageable pieces. One way is to use the ‘4Q method’. This comprises development qualification (DQ), installation qualification (IQ), operation qualification (OQ), and performance/process qualification (PQ). What the chosen manageable pieces or phases are is up to the individual, but it must be described in the validation plan. This defines the phases, the input and output of the phases, and which documents will be created during the phases. Typically, this will be a phase plan including test plans, the testing itself and the test documentation, and the phase report.
DQ – development qualification
This includes writing the user requirements specification, choosing the system, auditing the supplier if the risk assessment says that this is needed, and implementing the system.
IQ – Installation qualification
Installing the system is usually just a matter of following the description from the supplier. A brief test will show that the system is up and running.
OQ – Operation qualification
This may be defined differently in different organisations, but a common definition is to test each function separately. This is often what the supplier already has done, so the user may not have to do this if documentation and/or a supplier audit has shown that this has been done.
PQ – Performance or process qualification
The PQ is also defined differently in different organisations. Basically, the PQ is testing that the implemented system is according to business processes. This includes indirectly testing that the separate functions work as intended. This may also be called the system testing.
If the supplier’s OQ testing is unavailable, more of the functions may have to be included in testing. It is perfectly fine to combine the OQ and PQ into one combined phase.
It is important to qualify or validate all the functions needed for your workflows.
A thorough description of IT validation can be found in the book, International IT Regulations and Compliance. This book also has chapters on LIMS and instrument systems, and the tips there are useful to read and follow in order to get a really smart laboratory with the information required.
But validation is never done. It’s important to prove that the system is still validated, even after changes in and around the system. Having appropriate procedures to explain how to keep the validated state, and documentation to prove that procedures have been followed, are a must.
These procedures need to cover whatever is appropriate, including:
- Error handling, including corrective action and preventive action;
- Change management;
- Validation/qualification of changes;
- Backup and recovery;
- Configuration management;
- Disaster recovery and business continuity;
- E-signatures;
- Environmental conditions;
- Risk assessment and management;
- Security and user access;
- Service level agreements;
- System description;
- Training;
- Validation and qualification;
- Supplier audit;
- Daily use;
- Implementation of data in the system;
- Qualification/validation of implemented data in the system; and
- Data transfer between systems.
Some of these SOPs will be generic in the organisation, and some will be system specific.
Validation is a never-ending job, but with a validated system the user can be sure that the system works as intended and that the data is secured inside the system. That means that the user can prove beyond doubt that the data was entered on a given date and that the system will show that data has been corrected later.
Patent-related issues
The US patent system is based on ‘First to Invent’ and, in order to help determine who was first to invent, most companies engaged in scientific research create and preserve evidence that they can use to defend their patents at a future date. Traditionally, this evidence has been in the form of the bound paper laboratory notebook. In a patent dispute, any inventor is assumed to have an interest in the outcome of the case, so their testimony must be corroborated. Most organisations require these notebooks to be signed by the author (‘I have directed and/or performed this work and adopt it as my own’) and also by an impartial witness (‘I have read and understood this work’).[10, 11]

Evidence in US patent interferences is subject to the Federal Rules of Evidence. There are a number of important hurdles that need to be overcome, in particular the Hearsay Rule (by definition, if the author cannot be present, then the evidence is hearsay) and the Business Records Exception.
The Business Records Exception is an exception to the hearsay rule, which allows business records such as a laboratory notebook to be admitted as evidence if they can be demonstrated to be relevant, reliable and authentic. The following criteria must be met:
- Records must be kept in the ordinary course of business (e.g. a laboratory notebook);
- The particular record at issue must be one that is regularly kept (e.g. a laboratory notebook page);
- The record must be made by or from by a knowledgeable source (e.g. trained scientists);
- The record must be made contemporaneously (e.g. at the time of the experiment); and
- The record must be accompanied by testimony by a custodian (e.g. company records manager).
Any doubt about the admissibility of electronic records was largely removed by this statement from the Official Gazette (10 March 1998 [12]: ‘Admissibility of electronic records in interferences: Pursuant to 37 CFR 1.671, electronic records are admissible as evidence in interferences before the Board of Patent Appeals and Interferences to the same extent that electronic records are admissible under the Federal Rules of Evidence. The weight to be given any particular record necessarily must be determined on a case-by-case basis.’
In terms of admissibility, paper and electronic records are therefore equivalent. The judgment is made on the evidence, not the medium in which it is presented. However, it is important to understand the factors that impact upon the authenticity of electronic records and that in the adversarial nature of the courtroom, the opposing side may attempt to discredit the record, the record-keeping system, and the record-keeping process. The integrity of the system and the process used to create and preserve records are therefore paramount.
Many organisations still require their scientists to keep bound laboratory notebooks. This is because there isn’t the case law or other experience for most legal advisors to feel as comfortable with electronic records as they are with paper. The issue is not one of admissibility, but of the weight that the record will have in court. Unfortunately, we are unlikely to see a suitable body of case law for many years.
The high-stakes nature of the problem, lack of experience, and long-term accessibility concerns have caused a number of organisations to adopt a hybrid solution, using an electronic lab notebook (ELN) front-end tool to create records, and then preserving the resulting records on paper. This gives the benefits of paper records (for the lawyers) while providing the scientists with the benefit of new tools. A fully electronic system will require scientists to sign documents electronically, and the resulting record to be preserved electronically.
Using multiple systems for patent evidence creation and preservation can expose an organisation to increased risk, due to the need to maintain the integrity of each system, and the consistency of the content between them. Similarly, the use of generic systems for such a task can increase discovery concerns and also increase the likelihood of problems. Further guidance should be sought from records management personnel and legal advisors within the organisation, in order to determine policy.
A recommended approach to help uncover and resolve legal/patent concerns is to work with the company’s lawyers and patent attorneys to simulate the presentation of ELN evidence in the courtroom, and then work back to the creation of that evidence in the laboratory.
The America Invents Act – implications
Patent-reform legislation, in the form of the Leahy-Smith America Invents Act 2011, changed the US system from First to Invent to First to File in March 2013. It is very tempting to view this change as an opportunity to relax some of the procedural requirements of ELNs used in research laboratories.
However, there are clauses in the Act that would suggest it’s wise not to make such an assumption. It is likely that patent interferences and interfering patent actions will continue for many years for patents and applications filed after March 2013. [13]
There are specific circumstances described in the America Invents Act that, for example, require proof of inventive activities to remove prior art for joint research activities, or to preserve the right to an interference if the application contains, or contained at any time, a claim to an invention filed before March 2013. Until the act becomes effective, and there is clarification about the implications of the new legislation, there is no reason to change in-house procedures for keeping laboratory notebooks, or for vendors to revise the procedures and workflows in their ELN products. The more immediate concerns are:
- There is a loophole that will allow people to prosecute a patent under the old First to Invent rules for many years to come. First to File isn’t dead even after 16 March 2013 – there are some changes that mean proof of inventive activities will be especially important for joint research activities. The retention of other documentation related to joint research projects may need to improve; and
- Derivation proceedings will also require proof of inventorship.
To add further uncertainty, there’s always a chance (or indeed probability) that things are going to end up in the US Supreme Court to examine the constitutional implications of a move away from First to Invent. So it does appear that the new Act makes legally robust, signed, and witnessed records of inventive activities (generally in the form of lab notebooks) even more critical. With a move to ‘First to File’ there’s the additional pressure of getting to the Patent Office quickly, which means it is necessary to start paying attention to the patent filing process, which has historically not been under much time pressure.
Data integrity, authenticity and management
Whenever electronic records are used within the framework of legal or regulatory compliance, data integrity and data authenticity are fundamental requirements of the computer systems used to create, manipulate, store and transmit those records. These requirements may also apply to in-house intellectual property (IP) protection requirements. It will therefore be necessary for a laboratory informatics implementation project to very carefully consider the specific requirements of their organisation in this area. [14]
The characteristics of trustworthy electronic records are:
- Reliability – the content must be trusted as accurate;
- Authenticity – records must be proven to be what they purport to be, and were created and transmitted by the person who purports to have created and transmitted them;
- Integrity – must be complete and unaltered, physically and logically intact; and
- Usability – must be easily located, retrieved presented and interpreted.
Data integrity, in a general sense, means that data cannot be created, changed, or deleted without authorisation. Put simply, data integrity is the assurance that data is consistent, correct and accessible. Data integrity can be compromised in a number of ways – human error during data entry, errors that occur when data is transmitted from one system to another, software bugs or viruses, hardware malfunctions, and natural disasters.
There are many ways to minimise these threats to data integrity including backing up data regularly, controlling access to data via security mechanisms, designing user interfaces that prevent the input of invalid data, and using error detection and correction software when transmitting data.
Data authenticity is the term used to reinforce the integrity of electronic data by authenticating authorship by means of electronic signatures and time stamping.
Generally speaking, electronic signatures are considered admissible in evidence to ensure the integrity and authenticity of electronic records. An electronic signature is a generic term used to indicate ‘an electronic sound, symbol or process attached to or logically associated with a record, and executed or adopted by a person with the intent to sign the record.’
A digital signature is a specific sub-set of an electronic signature that uses a cryptographic technique to confirm the identity of the author, based on a username and password and the time at which the record was signed. The requirements for an informatics project will be somewhat dependent on the nature of the organisation’s business and internal requirements, but security, access control and electronic signatures are factors that must be given appropriate consideration.
There are a number of ways to ensure data integrity and authenticity. The first is to develop clear, written policies and procedures of what is expected when work is carried out in any laboratory; the integrity of the data generated in the laboratory is paramount and must not be compromised. This is the ‘quality’ aspect of the quality management system (QMS) that must be followed.
There is the parallel need to provide initial and ongoing training in this area. The training should start when somebody new joins the laboratory, and should continue as part of the individual’s ongoing training over the course of their career with the laboratory.
To help train staff, we need to know the basics of laboratory data integrity.
The main criteria are listed below:
- Attributable – who acquired the data or performed an action, and when?
- Legible – can you read the data and any laboratory notebook entries?
- Contemporaneous – was it documented at the time of the activity?
- Original – is it a written printout or observation or a certified copy thereof?
- Accurate – no errors or editing without documented amendments;
- Complete – all data including any repeat or reanalysis performed on the sample;
- Consistent – do all elements of the chromatographic analysis, such as the sequence of events, follow on and are they date- or time-stamped in expected sequence?
- Enduring – they must not be recorded on the back of envelopes, cigarette packets, or the sleeves of a laboratory coat but in laboratory note books and/or electronically by the chromatography data system and LIMS; and
- Available – for review and audit or inspection over the lifetime of the record.
It is important that laboratory staff understand these criteria and apply them in their respective analytical methods regardless of working on paper, hybrid systems or fully electronic systems. To support the human work, we also need to provide automation in the form of integrated laboratory instrumentation with data handling systems and laboratory information management systems (LIMS) as necessary. In any laboratory, this integration needs to include effective audit trails to help maintain data integrity and monitor changes to data.
Supervisors and quality personnel need to monitor these audit trails to assess the quality of data being produced in a laboratory – if necessary a key performance indicator (KPI) or measurable metric could be produced.
Chapter summary
From a broader business perspective, the introduction of computerised tools for managing laboratory information comes at a perceived higher cost, and challenges the user to consider very carefully the consequences of moving from a paper-based existence to one based on technology.
The return on investment equation is critical in obtaining the initial go-ahead for an informatics project, but the transition to digital from paper represents a major upheaval to long-established and well-understood information management processes.
Computer systems used in regulated environments need to be validated; the user needs to be confident that computerised systems can deliver productivity benefits, and data integrity and data authenticity can be guaranteed in a digital world.
Lawyers and patent attorneys need to be confident electronic lab notebooks can be presented as evidence in patent submissions, interferences and litigation.
Next: Practical considerations: Specifying and building the smart laboratory >
