
SYNTHIATM retrosynthesis software has become a game changer for drug discovery. Using expert-coded rules and AI-powered algorithms developed over 15 years, the software helps organic chemists develop robust synthetic routes to their target molecule.
Customised synthesis pathways can be designed using commercially available starting materials, building blocks, synthons, or a proprietary inventory. Moreover, results can be refined and optimised based on users’ preferences and project objectives.
Now, the team behind SYNTHIA has developed a sophisticated application programming interface (API) that can incorporate a wide range of cheminformatics tools, allowing users to streamline retrosynthetic analysis for promising leads and assess synthetic accessibility scores (SAS) of thousands of virtual molecules.
Synthetic accessibility score
Traditionally, combinatorial chemistry and generative modelling are used to construct vast compound datasets[1]. However, the actual synthesis of molecules obtained through such methods is challenging. The SYNTHIA Synthetic Accessibility Score (SAS) application programming interface offers a breakthrough solution. By combining deep-learning models with data from SYNTHIA retrosynthesis software, it provides predictions for molecule feasibility and complexity in terms of the number of synthetic steps, starting from small, commercially available building blocks. The machine-learning model behind SAS was pre-trained on synthetic scenarios from the SYNTHIA Retrosynthetic Planning Tool [2];[3];[4]. The solution offers the ability to easily process millions of molecules daily, enabling unprecedented speed in pathway design.
User dataflow
SYNTHIA SAS is an ISO-27001-certified, cloud-hosted service, which is available to customers via the RESTful API. It is horizontally scalable and provides high-throughput capability via a single API entry point for all customers. Moreover, the service is stateless and designed to scale as needed. Essentially, users can enter their molecules individually or as a batch in the SMILES text format [5], and the software returns a score for each molecule (Fig. 1).
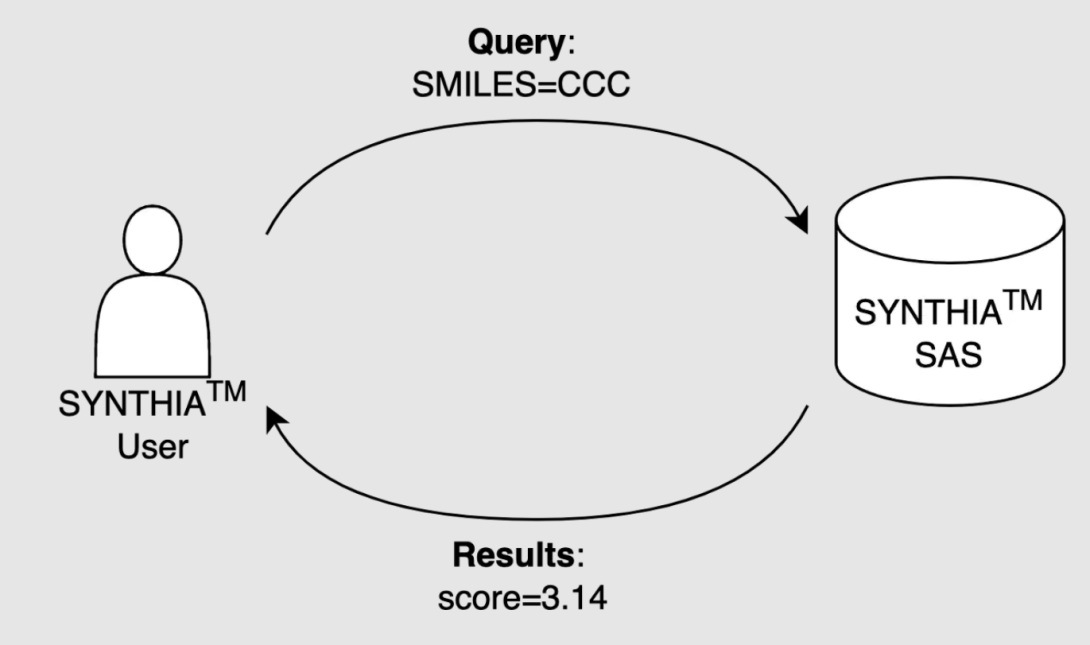
Figure 1. Schematic representation of SYNTHIA SAS service dataflow.
Predictive model characteristics
SYNTHIA SAS is based on a regressor that includes a Graph Convolutional Neural Network (GCNN). The architecture allows learning internal representations of molecules through their graph structures rather than pre-computed molecular descriptors[6]. The model consists of a bond-level directed message-passing neural network (D-MPNN) followed by a feedforward neural network (FNN), adapted from the Chemprop open-source project [7].
The machine-learning model was trained using SYNTHIA automatic retrosynthesis module results as target values. Specialised and normalised SYNTHIA scores were used to reflect the number of steps. Furthermore, only small building blocks were selected as search settings. A smoothing function was also applied to enhance the gradient for high scores and improve the resolution of molecules that are difficult to synthesise.
Case Study
N-acetyl derivative of sulfamethoxazole (Fig. 2, left) is a direct precursor of the drug sulfamethoxazole (Fig. 2, right). Despite its more complex chemical structure, the derivative is recognized as being easier to synthesize (SAS = 1.038 is much smaller than SAS = 4.051).
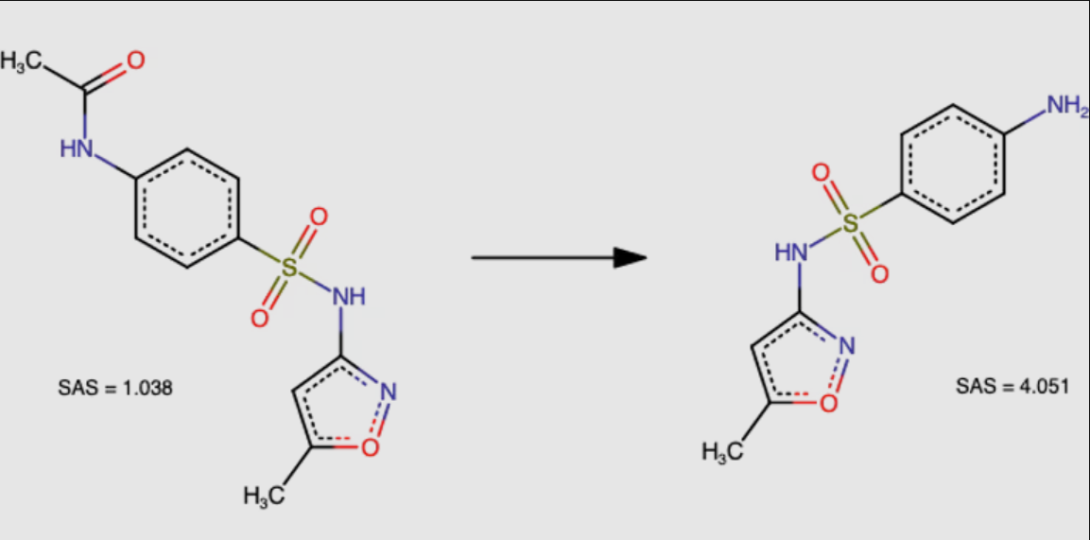
Figure 2. Chemical structures of molecules for sulfamethoxazole use case
Interpreting accessibility scores
For every molecule entered, the software generates a synthetic accessibility score (SAS) ranging from 0 to 10. This value approximates the number of steps required to synthesise the molecule using commercially available building blocks. The lower the score, the easier it is to synthesise the molecule.
Summary
Deciding whether a drug molecule is easy or difficult to synthesise is essential for streamlining virtual screening pipelines, but it is also very challenging and time-consuming. SYNTHIA SAS API offers a breakthrough solution by combining deep-learning models with data from SYNTHIA retrosynthesis software to enable high-throughput in-silico compound processing. The service allows organic chemists to analyse thousands of pathways in minutes, thus greatly accelerating and augmenting molecule selection prior to synthesis of active pharmaceutical ingredients.
www.synthiaonline.com
References
1. Joshua Meyers, Benedek Fabian, Nathan Brown. De Novo Molecular Design and Generative Models, Drug Discovery Today, 26, 2021, 2707-2715. DOI
2. www.sigmaaldrich.com/SYNTHIA
3. Tomasz Klucznik, et al. Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 4, 2018, 522-532. DOI
4. Mikulak-Klucznik, B., et al. Computational Planning of the Synthesis of Complex Natural Products, Nature, 588, 2020, 83–88. DOI
5. Daylight Chemical Information Systems, Inc.
6. Yang, K., et al. Analyzing Learned Molecular Representations for Property Prediction, Journal of Chemical Information and Modeling, 59, 2019, 3370-3388. DOI
7. github.com/chemprop/chemprop
